
家里蹲化学学会本科生Yunlong Shang在Nanoscale发表文章(Nanoscale, 2022, Advance Article),对先前报道的一例硼-锌团簇中Zn为+3氧化态的论文进行评论(Nanoscale, 2021,13, 14041-14048),指出该文章理论计算存在的不足之处。

在Jiawei Xu的指导下,他们结合多种波函数分析手段,辅以能量分析的角度,指出两个硼-锌团簇中的Zn仍然是+2氧化态。
首先使用了半经验LOBA(localized orbital bonding analysis)方法,对锌原子进行氧化态计算,在50%-90%的阈值范围内,团簇中的Zn均呈现+2氧化态。测试了不同定域化方法和布居计算方法对结果的影响,均得到了完全一致的结果。
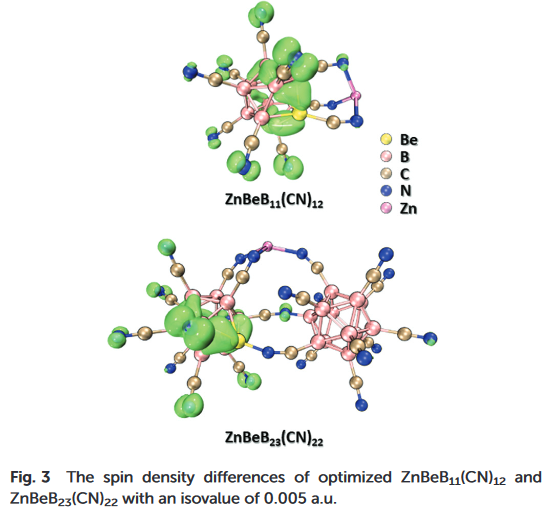
在这类具有净自旋密度且不涉及高自旋金属原子的体系中,自旋密度和自旋布居的角度往往可以很简便快捷地得到氧化态的结果。自旋密度差图表明,单电子成分出现在硼簇上,而锌原子上不存在未成对电子,即锌原子保持着d10的壳层结构,自旋布居计算也得到了完全一致的结果。
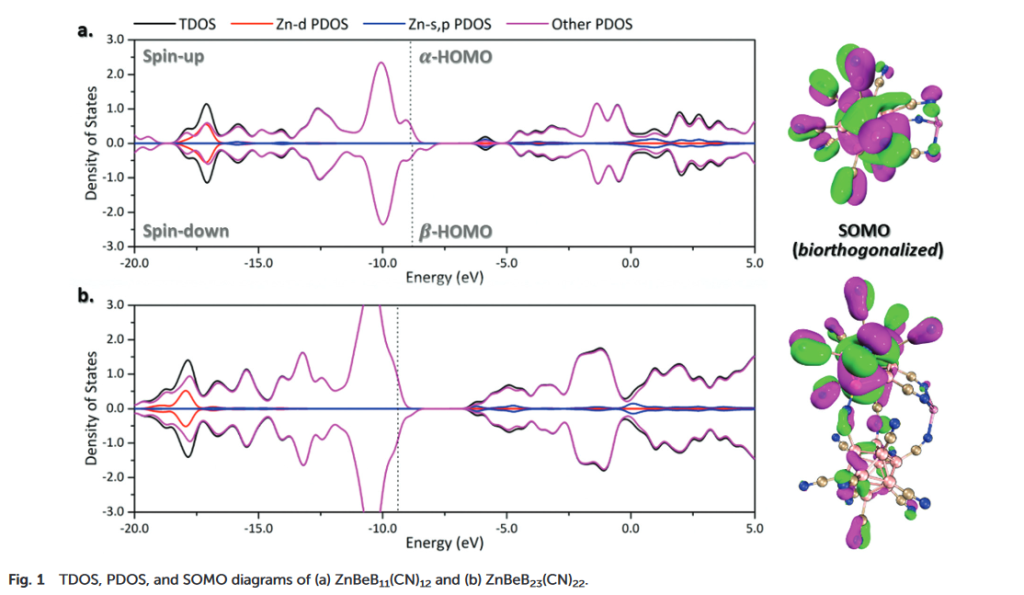
在态密度(DOS)图中,α电子和β电子的贡献基本上是对称的。但在接近HOMO能级的区域,可以观察到明显的单电子特征,这些不对称的部分中并没有锌原子任何轨道的贡献,而完全是由硼簇贡献的,同样可以得到Zn呈+2氧化态的结论。
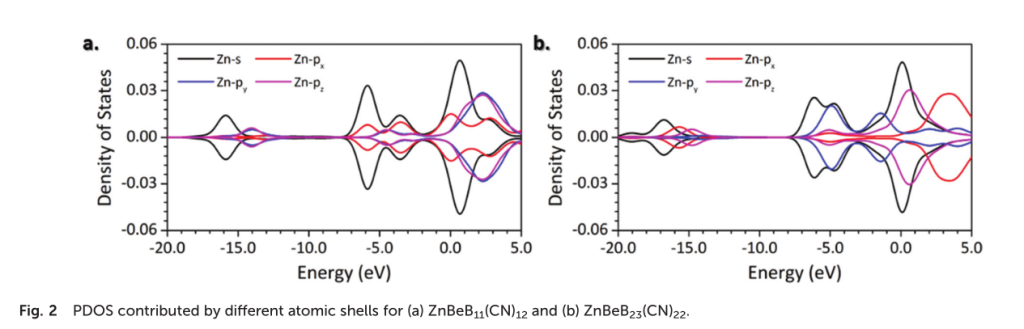
根据局部态密度图(PDOS)还可以用来分析锌原子的杂化特征:在占据成键区域内,锌原子的两个p轨道的贡献是相似的,而另一个p轨道的贡献是截然不同的,可以得到锌原子采取sp2杂化的结论。
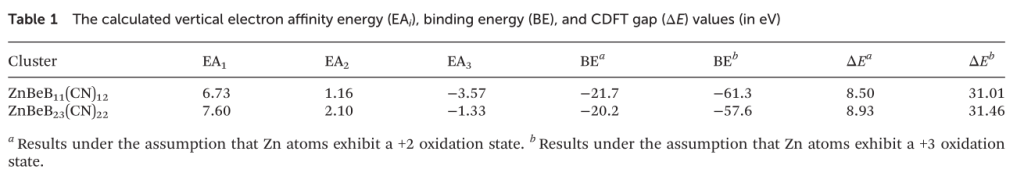
除了波函数分析的角度,能量的角度也可以用来判断氧化态。其中,较为常用的是计算结合能的方法与限制性DFT(CDFT)的方法,这两种方法的思路有一定的相似性,即都需要引入对氧化态的预设,如本文中Zn可能的氧化态为+2或+3,然后通过将计算得到的能量与DFT结果进行对比,从而判断哪一种设定更接近真实情况,即哪一种氧化态的结果更为合理。在本文的两个体系中,两种方法都指向Zn呈+2氧化态。还需要注意的是,原文中采取计算电子亲和能来估算氧化态的做法并不一定可靠,电子亲和能本身并不能衡量得电子后的结构是否真的稳定。
此外,通过理论计算的光电子能谱与实验光谱进行对比也可以用来说明团簇中锌原子的氧化态。他们还通过ETS-NOCV(extended transition state-natural orbitals for chemical valence)分析,讨论了锌原子与硼原子簇之间的成键结构。
本工作得到了江苏省大规模复杂系统数值模拟重点实验室(NSLSCS)的支持。