
Supercell.py:从原胞.cif建模超胞.pdb
Supercell.py: From Unit Cell .cif to Super Cell .pdb
Jiawei Xu
Released: 2023-01-14 / Updated: 2023-01-14
扩胞是材料计算中时常会遇到的步骤。常见带有周期性体系建模功能的软件如GaussView、Vesta、Materials Studio、VMD等都可以进行扩胞操作。在分子动力学和QM/MM计算中,最常使用的结构文件格式是.pdb,这不能通过以上软件由.cif文件方便、直接地产生。笔者是建模的懒人,致力于以易用的GaussView为主完成各种需求的建模,因此本文将通过一个实例介绍Supercell.py这一小程序的使用方法,该程序可从本文结尾处链接下载。
我们从COD下载ID为4024905的晶体文件,这是一个BODIPY衍生物的结构。用GaussView打开可看到一个单胞中包含了取向不同的两个BODIPY分子,且已经包含了氢原子,共80个原子。首先用VASP对该结构进行简单优化,将所得CONTCAR用Vesta导出为.cif文件,结构和晶胞参数基本没有发生太大改变。在GaussView中打开优化后的结构,观察发现分子被周期性边界截断,这会导致后续建模时residue定义出现问题,也不能保证超胞边界上分子的完整性。
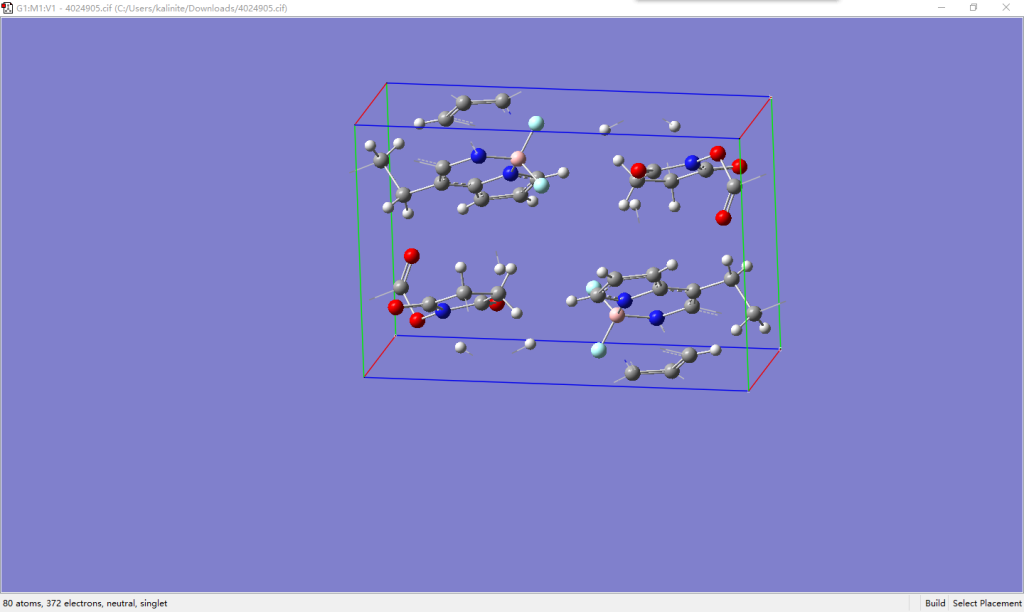
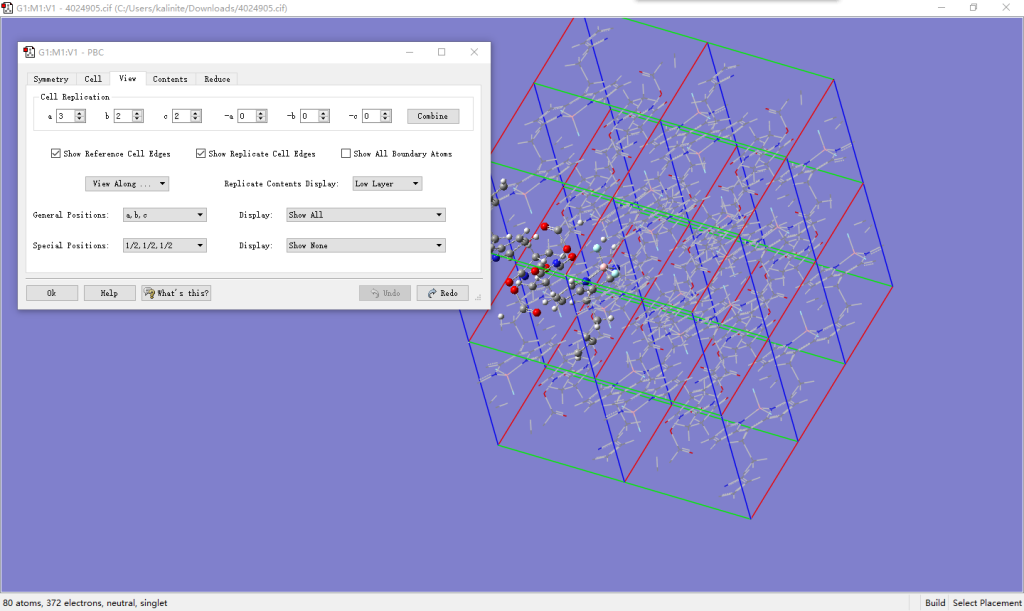
首先对其在GaussView中扩胞,构建3×2×2的超胞,此时保证了超胞中心有一对完整的分子,点击Combine。对于性能一般的笔记本此时会比较卡顿,但我们并不需要针对这个超胞建模。适当调整视角,用框选工具删除周围多余的分子,只保留中间一对取向不同的分子。删除一批后,由于原子数变少,转动会更为方便。保存最后的两个分子的坐标为.gjf文件。再次打开.cif文件,直接保存为另一个.gjf文件,然后将前面的坐标替换.cif导出的.gjf中的坐标,注意不要改动Tv的坐标,打开后可以看到此时的分子已经是完整的,用分层功能记录两个分子各自的原子序号。对于本例分别是1-26,41,44-56和27-40,42-43,57-80,操作不同可能有所区别。至此,建模游戏已经结束。
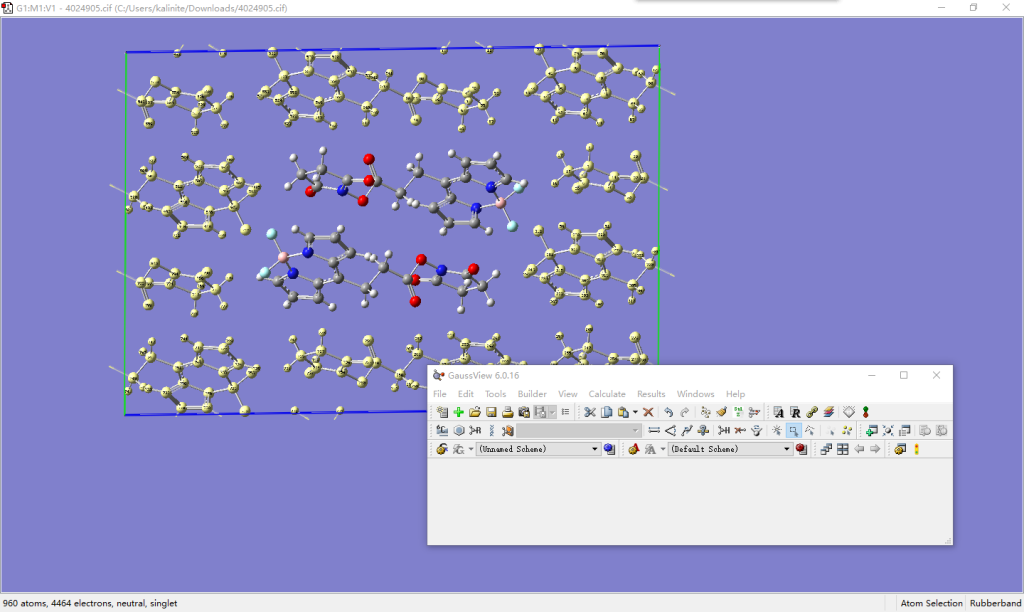
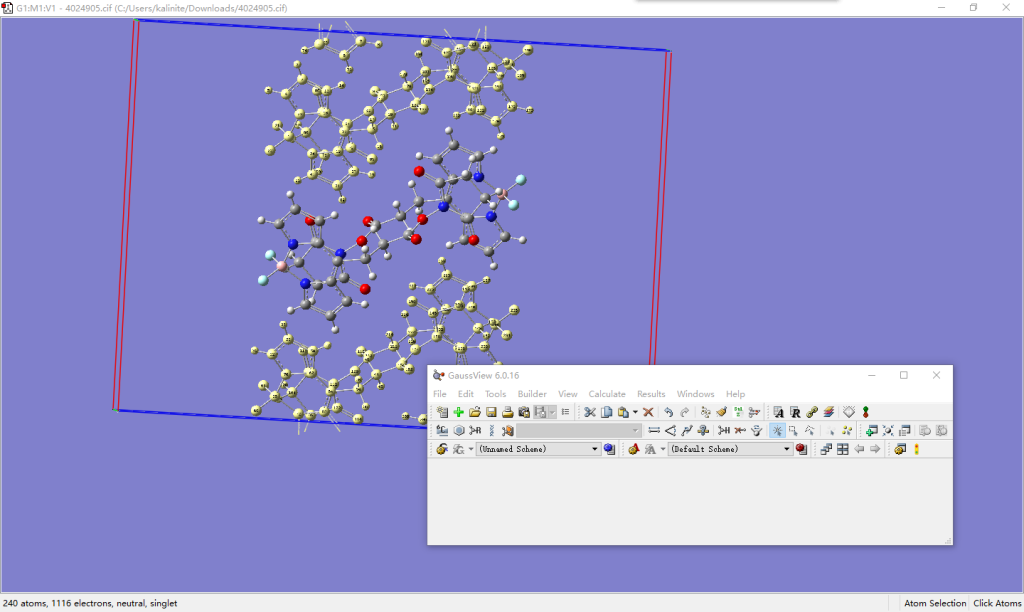
打开Supercell.py文件,修改文件开头extend和residues的值。extend中定义了向x+、x-、y+、y-、z+和z-六个方向扩胞的范围,实际上只要是整数均可,程序构建超胞时只关心二者的差值,若(0, 0, 0)包含在该范围内则会保留原始坐标下的单胞。如果你想构建二维的超胞,将某一方向的数值填为(0, 0)即可。residues中定义了残基包含的原子序号以及残基名,必须包含单胞中的所有原子,且不重不漏,残基名应保持在三个字符以内。
运行python Supercell.py 4024905.gjf 4024905.pdb。用VMD观看所得的.pdb文件。对于本例,我们扩建9×5×5的超胞,即设置extend的数值分别为(-4, 4)、(-2, 2)和(-2, 2)。程序运行时输出有关原胞和超胞大小的信息,并指出原胞在超胞中的残基号是225~226。这一信息可以帮助我们在VMD中配合.tcl脚本,通过选择语句导出核心区域周围一定范围的原子序号或残基号,进而用于做QM/MM计算。
对于需要构建有缺陷的超胞,也可以通过简单计算判断需要修改哪一个残基号,然后直接打开.pdb修改、或通过VMD选区保存后修改即可。